Atrofia muscular espinal
Es una enfermedad autosómica recesiva, causada por mutaciones en el gen SMN1, que conduce a una reducción de la expresión de la proteína de neurona motora de supervivencia (SMN por sus siglas en inglés: Survival Motor Neuron).
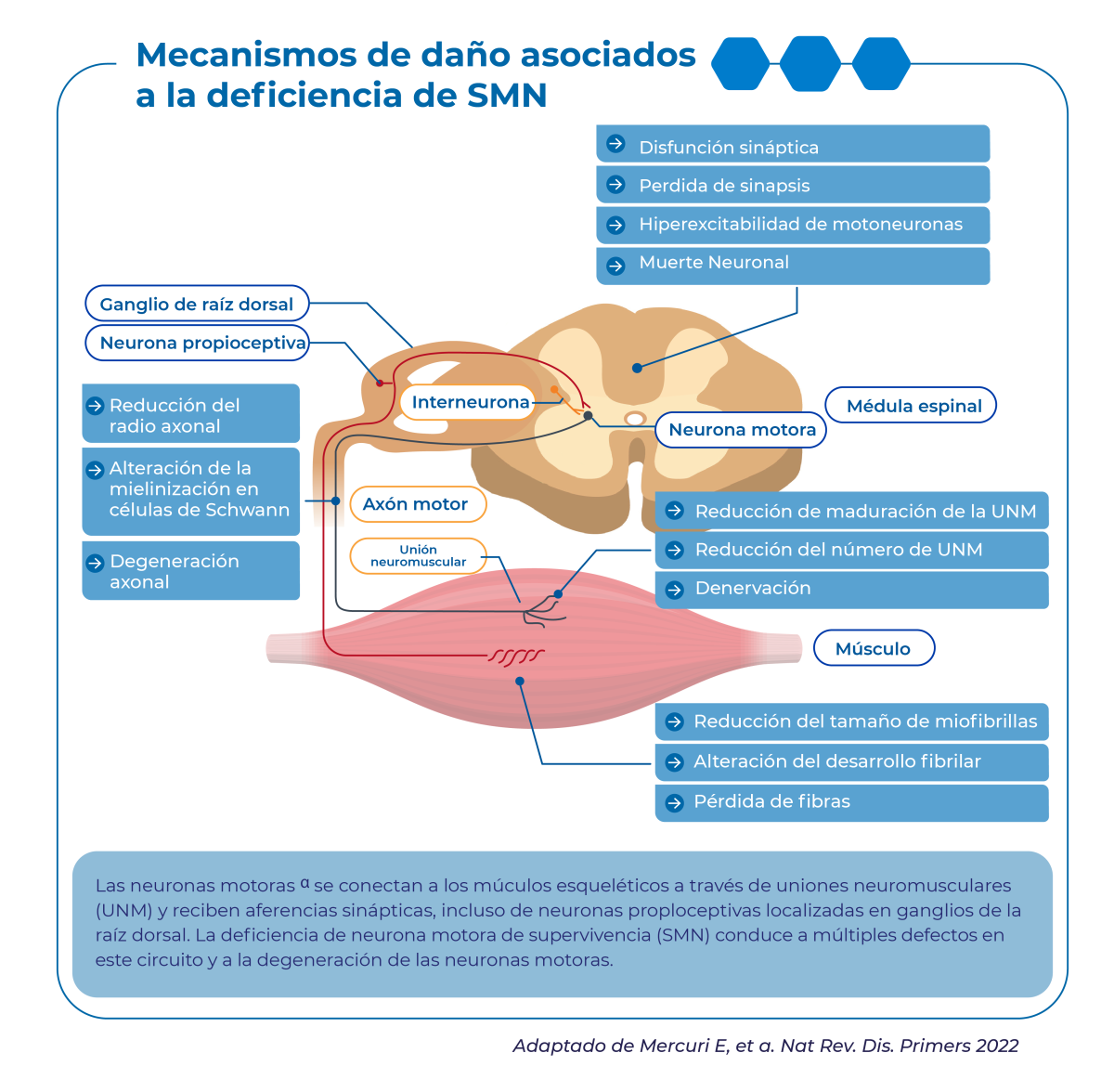
Esto provoca una degeneración de las α-motoneuronas en la médula espinal y tronco encefálico, asociada a hipotonía, atrofia muscular progresiva y debilidad de extremidades, tronco y músculos respiratorios/deglutorios (Mercuri et al., 2022, Ogino et al., 2002; Urrutia et al., 2020).
La incidencia mundial de la atrofia muscular espinal menudo se informa como 1 a 2 casos por cada 10,000 personas, con importantes diferencias entre países y una frecuencia de portadores promedio de 1 en 50 (Pearn et al., 1978). Mientras que en México se estima una cifra de 0.5 a 1 por cada 25,000 nacimientos.Urrutia


Otros pacientes portan una variación patogénica puntual en SMN1 y una deleción en el otro alelo de SMN1 o, muy raramente, mutaciones bialélicas en cualquiera de los exones de SMN1 (Wirth et al., 2021).
El gen SMN1 es el principal que codifica consistentemente la proteína SMN de longitud completa, que es esencial para la supervivencia de las neuronas motoras, en contraste con que solo el ~10 % de la proteína SMN producida por el gen SMN2 (también conocido como gen “de respaldo”) es funcional.
La alta variabilidad fenotípica en el espectro de la atrofia muscular espinal se atribuye principalmente a la variedad de copias del gen SMN2, al ser un gen homólogo al gen SMN1, excepto por algunos nucleótidos.Schorling El gen SMN2 tiende a presentar un empalme alternativo (alternative splicing) durante la transcripción del ARNm, lo que origina una proteína trunca, poco oligomerizante, y de rápida degradación, con solo el 10% de la proteína SMN completa.Cardona En efecto, numerosos estudios muestran cómo es que a mayor número de copias del gen SMN2, mayor cantidad de proteína SMN de longitud completa y menor severidad en el fenotipo de AME y viceversa. Sin embargo, esta correlación inversa no es absoluta (Calucho et al., 2018).

La proteína SMN tiene una función fundamental en todas las células del organismo y su ausencia total tiene efecto letal in uteroTizzano, ya que la expresión de la proteína SMN es mayor durante el desarrollo neuronal y la localización de la proteína cambia de la región nuclear a la citoplasmática en las neuronas maduras, también cumple funciones en el desarrollo y la maduración de las neuronas en la señalización y transmisión de la unión neuromuscular (Chaytow et al., 2018).

Manifestaciones clínicas
Aunque la AME es un continuo de expresión fenotípica, los síntomas de presentación difieren significativamente entre subtipos (ver tabla 1).
Entre las características comunes a todo el espectro se incluyen la neuropatía motora pura y la axonopatía con arreflexia y fasciculaciones, conservación de la sensibilidad y un patrón típico de debilidad muscular, que es más grave para los músculos proximales y músculos de las extremidades inferiores (Kizina et al., 2021). Cuando no se trata en su forma más grave (Tipo 1), la degeneración de las neuronas motoras inferiores comienza poco antes del nacimiento y aumenta rápidamente, lo que puede ocasionar el uso ventilación permanente o muerte antes de los dos años si no se recibe tratamiento.
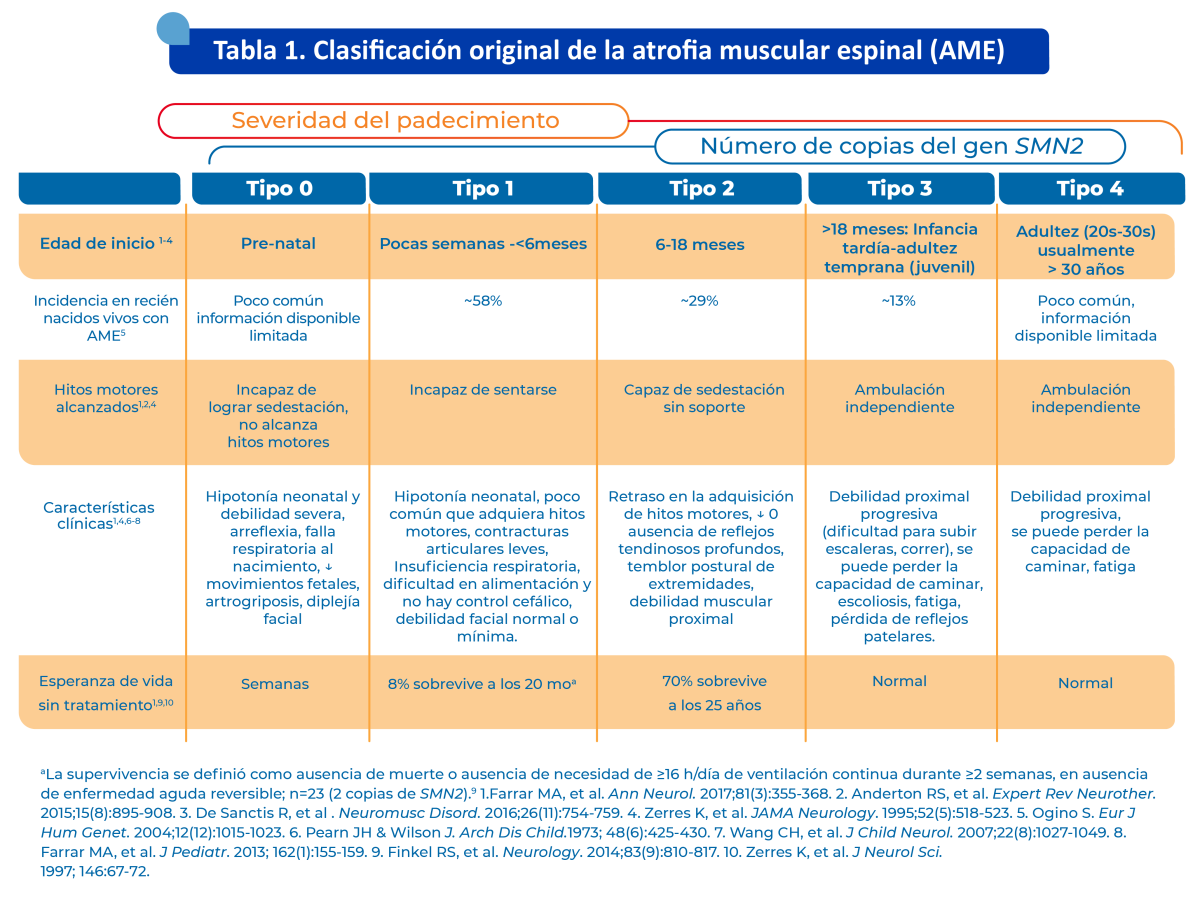
La atrofia muscular espinal se divide en 5 subtipos principales. El tipo 0, en el que solo existe una copia del gen SMN2, tiene presentación prenatal y una esperanza de vida de menos de 6 meses. Los pacientes presentan debilidad severa al nacimiento, hipotonía profunda, arreflexia, insuficiencia respiratoria, contracturas articulares y cardiopatía.Urrutia
El tipo I (enfermedad de Werdnig-Hoffman) es el más frecuente, sumando entre 50 y 70% de los pacientes. El genoma presenta un par de copias del gen SMN2 y en algunos casos se pueden presentar tres copias. Se presenta entre los cero y seis meses de edad e implica una esperanza de vida de menos de dos años, y el riesgo del uso de soporte respiratorio permanente. Se caracteriza por debilidad, hipotonía, fasciculaciones linguales, hiporreflexia o arreflexia, alteración de la succión y deglución e insuficiencia respiratoria.Urrutia
La atrofia muscular espinal de tipo II (enfermedad de Dubowitz) comprende a 20% de los pacientes, que poseen tres copias del gen SMN2. Esta variante se presenta entre los seis y 18 meses. Alrededor de 70% de los pacientes sobrevive a la edad de 25 años, aunque nunca presenta bipedestación ni deambulación. Otras características incluyen la debilidad proximal, hipotonía, temblor postural de manos, hiporreflexia y escoliosis.Urrutia


Complicaciones
Durante su evolución, aparecen distintas complicaciones entre las que destacan las deformidades articulares, escoliosis, luxación de caderas, fracturas y alteraciones respiratorias.Febrer
Las deformaciones articulares son una complicación frecuente y aparecen con mayor frecuencia en el tipo II de atrofia muscular espinal. Por otra parte, la escoliosis ocurre de forma muy frecuente, pues tiene una incidencia de entre el 78 y el 100% de los pacientes. En cuanto a las fracturas y contracturas, existe mayor riesgo debido a una importante presencia de osteoporosis por inmovilización y falta de actividad muscular.Urrutia
La principal causa de muerte en niños con atrofia muscular espinal son las alteraciones respiratorias, sobre todo en los tipos más graves de enfermedad. En el tipo III, este tipo de complicaciones son menos frecuentes.Urrutia
Diagnóstico
Históricamente, la electromiografía (EMG) y la biopsia muscular establecían el diagnóstico. Sin embargo, estas pruebas ya no se realizan de forma rutinaria y hoy en día son las pruebas genéticas el paso diagnóstico inicial.
Nuevas recomendaciones sugieren determinar también el número de copias de SMN2, ya que esto proporciona información sobre la gravedad de la enfermedad en pacientes sintomáticos e identifica qué pacientes presintomáticos pueden iniciar un tratamiento (Schorling et al., 2019).



Tratamiento
El cuidado de los pacientes con atrofia muscular espinal requiere del manejo interdisciplinario de los problemas respiratorios, nutricionales, gastroenterológicos, ortopédicos y psicosociales (Schorling).
Antes de 2017, el manejo de los pacientes era de apoyo y se recomendaban más intervenciones invasivas en los pacientes con mayor debilidad Friedman.
Pero ha habido avances en el campo terapéutico gracias al mayor conocimiento en cuanto a las bases moleculares de la enfermedad, lo que ha resultado en terapias avanzadas muy específicas.Tizzano